
Peter Doshi, editor of the British Medical Journal (BMJ) is once again bravely sticking his neck out with his latest co-authored paper.
He and six other authors from the US, Spain and Australia decided to investigate serious adverse events of special interest following mRNA vaccination in randomized trials.
They used a World Health Organisation (WHO) endorsed priority list of adverse events of special interest (AESIs) to identify serious adverse events that occurred during phase III clinical trials on which emergency use authorisation (EUA) was based. The list comprised of adverse events based on the specific vaccine platform, adverse events associated with prior vaccines in general, theoretical associations based on animal models and COVID-19 specific immunopathogenesis.
The study looked at the Pfizer and Moderna trial data which was expected to follow participants for two years. However, after EUA, participants were unblinded and those in the placebo arm offered the vaccine. This meant post authorisation data was less reliable so, to preserve randomisation, the authors used interim datasets from approximately 4 months after the trials commenced.
A serious adverse event (SAE) was defined as an adverse event that results in any of the following conditions: death; life-threatening at the time of the event; inpatient hospitalisation or prolongation of existing hospitalisation; persistent or significant disability/incapacity; a congenital anomaly/birth defect; medically important event, based on medical judgement.
SAE tables were located for both the Pfizer and Moderna trials submitted for EUA in the US. For the Moderna trial, these SAEs were from dose 1 but for Pfizer they were from dose 1 to 1 month after dose 2.
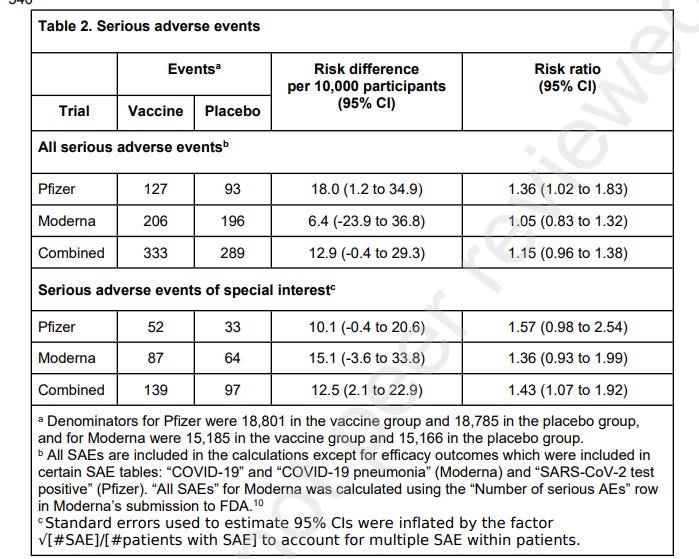
The authors found that, in the Pfizer trial, there was a 36% higher risk of SAEs, unrelated to COVID-19 in vaccinated participants in comparison with the placebo group. The Moderna trial had a 5% higher risk of SAEs. Combined there was a 15% higher risk of SAEs in the vaccine recipient group.
52 AESIs were reported in the Pfizer vaccine group versus 33 in the placebo group. This meant there was a 57% increased risk of serious AESIs and an absolute risk increase of 10.1 serious AESI per 10,000 vaccinated participants.
In the Moderna trial, there were 87 serious AESIs in the vaccine group versus 64 in the placebo group, representing a 36% increased risk. In this trial, there was an absolute risk increase of 15.1 serious AESIs per 10,000 vaccinees.
Combined, there was a 43% increased risk of serious AESIs and an absolute risk increase of 12.5 serious AESIs per 10,000 vaccinated participants.
In both trials, the largest increase in absolute risk was in coagulation disorders. There were more cardiovascular AESIs in the vaccine group in the Pfizer trial but it was balanced in the Moderna trial.
Taking into consideration the harm-benefits, they compared AESIs with vaccine risk reduction for COVID-19 hospitalisation. With the Moderna vaccine, the risk of AESIs was 15.1 per 10,000 participants compared with a Covid hospitalisation risk reduction of 6.4 per 10,000. The Pfizer trial had a AESI risk of 10.1 per 10,000 compared with a hospitalisation risk reduction of 2.3 per 10,000 participants. Therefore, with both vaccines, the risk of AESIs surpassed the risk reduction for COVID-19 hospitalisation.
The paper looks at the FDA reviews of SAEs which concluded that, for Pfizer, were “balanced between treatment groups”. For Moderna, the FDA said that SAEs were “without meaningful imbalances between study arms”. This current study, however, found an increased risk of SAEs so they tried to understand how there could be two very different conclusions. They concluded that the main reason for the discrepancy may be because the FDA used a different, larger analysis population. This was because the FDA included all individuals who had received at least one dose, irrespective of the duration of post-injection follow up time. The authors, in this paper, used a study population with medial follow up >2 months after dose 2, of which 98.1% had received both doses. Therefore, the FDA’S analysis included thousands of additional participants with very little follow-up, of which the large majority had only received 1 dose.
The authors discuss the results which show an excess risk of serious AESIs greater than the reduction in COVID-19 hospitalisation. They say that these results are compatible with another analysis (Benn et al) which found no evidence of a reduction in overall mortality. They say that both papers point to the need for formal harm-benefit analyses especially in individuals at low risk of COVID-19 hospitalisation or death.
It is suggested that because the majority of SAEs are relatively common events (e.g. stroke), signal detection is complicated because clinical suspicion of adverse vaccine reactions will be lower than less common SAEs such as myocarditis. Therefore, there is likely to be over and under reporting. Public health messages assuring safety may lower clinical suspicions whereas messages about potential harms may stimulate reports.
Because this study only looked at SAEs occurring past 1 month after dose 2 there may have been undercounting of serious AESIs.
The authors conclude that a systematic review and meta-analysis using individual participant data should be undertaken to address questions of harm-benefit in various demographic subgroups. Full transparency of the COVID-19 vaccine clinical trial data is needed to properly evaluate these questions. Unfortunately, well over a year after widespread use of COVID-19 vaccines, participant level data remain inaccessible.
Source: nakedemperor.substack.com/p/editor-of-the-british-medical-journal-6d1?utm_source=email
2 thoughts on “Editor of the British Medical Journal, Peter Doshi’s latest paper shows COVID-19 vaccines associated with increased risk of serious adverse events.”